Obesity, a major public health concern, increasingly impacts key physiological systems, including respiratory health.
Obesity Hypoventilation Syndrome
-
- Updated:
- 20 minute read
- 0 Comments
Obesity is a major public health issue and a topic of passionate debate through the 21st century. The increasing trend of obesity and emergence of super obese individuals (body mass index [BMI] 50 kg/m2) has led to a plethora of health consequences affecting major physiological and biochemical systems including the respiratory system (Finucane et al. 2011).
In absence of lung and neuromuscular disease, the development of awake- hypoventilation and consequent hypercapnia (PaCO2. 45 mm Hg) in obese patients (BMI. 30 kg/m2) is referred to as obesity hypoventilation syndrome (OHS). OHS patients display numerous abnormalities when subjected to a polysomnography test, including obstructive sleep apnoea (OSAS) with hypercapnia and limited airflow causing obstructive hypoventilation during sleep stages (Hart et al. 2013).
The presence of daytime hypercapnia is a key defining factor in OHS. However, there are obese individuals that do no develop awake hypercapnia despite hypoventilation during sleep, this observation may indicate the presence of a prodromal phase in development of OHS, which ultimately manifests in awake hypercapnia (Lavie 2008). In addition, there is a positive correlation between increasing obesity and incidence of OHS. Studies have shown that up to 20% of outpatients in sleep clinics and approximately half of all patients with a BMI of 50 kg/m2 and above may have OHS (Sotos 2003).
Here, we will look at the means by which awake hypoventilation may develop in patients with obesity, whilst also focusing on the pathophysiology and the clinical manifestations of OHS. In addition, we highlight recent data emerging from studies assessing the efficacy of various methods of therapy for OHS and attempt to review current and potential screening methods for this condition whilst highlighting the importance of early detection in this debilitating and costly condition.
2. Diagnosis of OHS & its clinical presentation
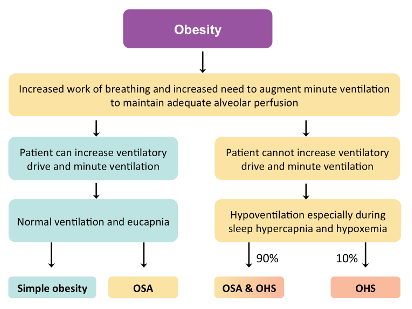
Figure 1 | A simplified algorithm illustrating the pathophysiology of obesity hypoventilation syndrome (OHS) and obstructive sleep apnoea (OSA) (Adapted from Laila Al Dabal 2005)
Repeated hospitalisation is a common occurrence amongst OHS patients. Such patients usually present with acute respiratory acidosis in an episode of acute on chronic exacerbation, which often leads to intensive care unit admission (Teichtahl 2001). Severe obesity (BMI > 40kg/m2) is a common finding in OHS patients often accompanied with severe sleep apnoea (based on the Apnoea–Hypopnoea Index) and hyper-somnolence (Quint et al. 2007).
There are many shared similarities in terms of symptoms between OHS and OSA, such as snoring, reduced daytime wakefulness, witnessed apnoea and morning headaches. However, distinguishing factors such as increased burden of dyspnoea and signs of cor pulmonale in OHS sets them apart from uncomplicated OSA patients (Kessler et al. 1996). On physical examination, obesity is a common finding in OHS, accompanied by an increased neck circumference, enlarged uvula and tonsils, as well as oedema in the lower extremities. In addition, although difficult to hear, the pulmonary component of the second heart sound appears more noticeable in patients with OHS (Mokhlesi et al. 2007; Resta et al. 2000).
Arterial blood gas measurements remain the standard definitive test for identifying alveolar hypoventilation. The ongoing metabolical compensation of respiratory acidosis in chronic hypercapnia, present in OHS, leads to raised serum bicarbonate levels (Nowbar et al. 2004; de Llano et al. 2005). A combination of serum bicarbonate levels, severity of obesity and OSA can provide us with a physiologically sensible and less invasive, yet readily available test that can screen for hypercapnia with an acceptable degree of accuracy.
Daytime hypoxemia is far more common in OHS patients than in those with simple OSA. Thus, wakefulness measures of arterial oxygen saturation using tests such as finger pulse oximetry can aid in identification of OHS in groups of patients with OSA. Furthermore, sleep periods spent with oxygen saturation of less than 90%, which can be detected by polysomnography, can also be useful in detection of OHS (Weatherall & Beasley 2011).
Pulmonary function testing and chest imaging are helpful in identifying extra causes of hypercapnia, other than hypoventilation. OHS patients can present with normal pulmonary function tests; however, due to their body habitus, they often exhibit restrictive defects of mild to moderate severity with normal or close to normal ratios of FEV1/FVC. Overall, it appears that OHS patients retain better lung functions than those affected by other causes of hypercapnia (Mokhlesi 2010).
Severe obesity in OHS patients drastically limits their expiratory reserve volume. OHS patients may also exhibit mild reductions in maximum expiratory and inspiratory pressures, which is likely to stem from altered respiratory mechanics and changes in respiratory muscle strength (Janssens et al. 2003).
3. Obesity and respiratory mechanics (pathophysiology)
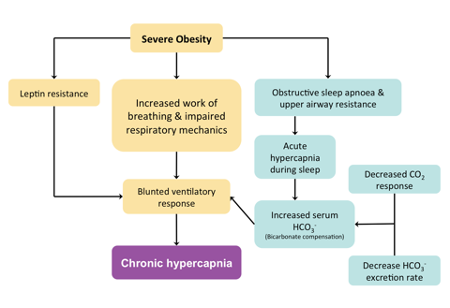
Figure 2 | An outline of the interactions between mechanisms which can lead to chronic daytime hypercapnia in patients with severe obesity (Adapted from Mokhlesi 2010)
The detrimental effects of obesity on pulmonary mechanics and respiratory muscle performance are well documented. However, when comparing obese patients, those suffering from OHS display augmented changes in their respiratory mechanics (Nowbar et al. 2004).
A reduction in compliance of the chest wall and the respiratory system, as well as an increase in airway resistance can occur as a consequence of breathing at abnormally low lung volumes (Spritzer 2004). Having an abnormally low exploratory reserve volume can lead to expiration flow limitation due to closure of smaller airways and air trapping, thus manifesting in an intrinsic positive end – expiratory pressure (Laaban & Chailleux 2005). Such alterations are more prominent in the supine position where the inspiratory muscles have to withstand an increased load (Mokhlesi et al. 2012). When comparing obese patients, those with OHS have been shown to significantly work harder for breathing regardless of whether they’re sitting or are in the supine position, awake or sleep (stage II sleep) (Salord et al. 2013).
One of the reasons for the augmentation of impairments seen in the respiratory function of OHS patients may stem from the debilitating effects of biomedical disturbances linked to hypoventilation, namely acidosis and hypoxemia. The existence of a primary myopathic process is also suggested (Mokhlesi et al. 2012), but due to lack of detailed studies on the muscle structure of OHS patients this topic remains highly speculative.
Studies exploring the mechanism behind the amplified reduction in respiratory function in OHS have demonstrated that patients with OHS have a larger neck circumference and a waist to hip ratio of greater scale in comparison to obese patients who are eucapnic or have OSAS (Golpe 2002).
The upward displacement of the diaphragm in central adiposity is associated with reduced mechanical performance, which is exacerbated in the supine position (Akashiba et al. 2006). Additionally, the lung volume is also negatively affected by central fat distribution (Mokhlesi et al. 2007), leading to a marked reduction in volume and increased mechanical ventilator limitations, thus increasing the effort required for breathing, while at the same time deteriorating the efficiency of the respiratory muscles in OHS. Furthermore, gas exchange is believed to occur less optimally in individuals with central obesity, irrespective of their BMI or sex (Kaw et al. 2009).
3.1 Upper airway obstruction
In majority of OHS patients, upper airway obstruction is an underlying condition that influences the emergence of awake-hypoventilation (Nowbar et al. 2004). Large proportions of OHS patients display upper airway obstruction when subjected to polysomnography, and often report a past history of snoring and apnoea (Emirgil & Sobol 2015). Daytime sleepiness and reduced concentration also affect OHS patients, which is a characteristic feature of OSAS. Continuous positive airway pressure (CPAP) therapy has proven to alleviate such symptoms and helps prevent OSA in OHS patients with sleep hypoventilation (Masa et al. 2001).
Given the lack of large longitudinal studies at present focusing on OHS development over time, it is possible that OHS could occur spontaneously in some individuals without upper airway dysfunction. In such individuals the rise of hypercapnia has been shown to be independent of the apnoea-hypopnea index (AHI) (Sidney Burwell et al. 1956); A proportion of researchers believe that the duration of disordered breathing events is more important in determining the severity and development of hypercapnia, rather than the frequency of the events occurring during sleep (Mokhlesi et al. 2012). In this context, acute increases in CO2 is associated with long periods of abnormal breathing and relatively shorter periods of restorative ventilation, whereby respiratory efforts may also be reduced due to underlying problems such as obesity (Mokhlesi et al. 2006).
A current hypothesis has linked the onset of chronic awake hypercapnia with the gradual retention of CO2 during sleep disordered breathing events. It is proposed that the rise in renal bicarbonates for buffering acute nocturnal rises in CO2eventually results in a less responsive ventilatory drive to CO2 during awake periods, ultimately manifesting in daytime hypoventilation (Piper & Grunstein 2010).
The emergence of awake hypercapnia is dependent on a combination of factors, including a compromised renal compensatory mechanism, dampened central respiratory drive and abnormal central obesity (Freedman 2002). Since these risk factors are subject to great variance amongst patients suffering from OSAS, the above hypothesis can help us understand why only certain groups of individuals display awake hypercapnia.
Furthermore, hypercapnia has been shown to develop when there is sustained hypoxemia (Oxygen saturation of less than 90%). Whether hypoxia is the instigating factor or the consequence of hypercapnia in the affected individuals is yet inconclusive, however, since the synthesis of certain neurotransmitters involved in central respiratory control can be negatively affected by hypoxia, it may suggest a causative role for hypoxia (Sakakibara et al. 1999).
3.2 Respiratory muscles
OHS patients display a reduction in maximal inspiratory and expiratory pressures (BECKER et al. 1999). In order to gain a better understanding of the efficacy of respiratory muscles in OHS, especially of the diaphragm, there is a necessity for more detailed investigations, such as cervical magnetic stimulation for testing diaphragmatic strength in OHS patients. However, since patients with OHS can generate trans-diaphragmatic pressures similar to that of eucapnic obese subjects at any level of diaphragmatic activation, casts a shadow of doubt on the importance of diaphragmatic weakness in the pathogenesis of this disorder (Society 2002).
3.3 Blunted Central Respiratory drive
There is a growing body of evidence that highlights the defectiveness of the central respiratory drive in OHS. The fact that OHS patients can reach eucapnia simply by voluntarily hyperventilating is a testament to this notion (Leech et al. 1991). There is a difference in hyperventilation between OHS patients and those suffering from morbid obesity, such that OHS patients do not hyperventilate as much (when rebreathing CO2), a defect that responds positively to CPAP therapy (Lin 1994). In addition, the increase in minute ventilation of OHS patients is not as extensive as when they’re subjected to breathing a mixture of hypoxic gases, indicating a blunted hypoxic drive, which is also amenable to CPAP therapy (Han et al. 2001), thus suggesting that such complications are most likely a secondary consequence of the syndrome.
Genetic predisposition, obesity, sleep apnoea and resistance to leptin have all been indicated in mechanisms that may lead to a blunted response to hypercapnia. Increased weight load was thought to be a direct cause of rising PaCO2 levels in OHS based on observations that weight loss results in improved PaCO2 in these patients, however it is unlikely that there is direct link between weight load and hypercapnia, since contrasting studies have shown that weight loss blunts the response of the eucapnic morbidly obese patient to hypercapnia (Emirgil & Sobol 2015).
A study comparing the ventilator response of first-degree relatives of OHS patients and control subjects has concluded that the existence of a familial genetic link in the blunted respiratory response to hypercapnia is unlikely, as both groups were shown to have similar ventilator response to hypercapnia (Jokic et al. 2000).
3.4 Leptin
Amongst the few number of hormones that stimulate ventilation, leptin, a satiety hormone produced by adipocytes is of particular interest. The excessive adipose tissue in obese patients leads to increased levels of leptin production; this serves to reduce the obesity related rise in CO2 load by increasing ventilation (Phipps et al. 2002). This mechanism helps explaining the absence of hypercapnia in majority of obese individuals.
Leptin levels are markedly higher in OHS patients when compared to healthy BMI matched individuals (CHERNIACK et al. 2008). However, it remains to be explored as to whether there is a direct link between OHS and elevated leptin levels; currently, the excess adipose tissue is believed to be the primary instigator of this increase in leptin levels. Furthermore, investigations into serum leptin levels of OHS patients have shown a significant increase in levels of this hormone when compared to body fat matched subjects with OSA who are eucapnic (Tankersley et al. 1998). The decline in serum leptin levels after PAP in these patients could be an indication of leptin resistance. When comparing the leptin cerebrospinal fluid (CSF) to serum ratio in healthy lean individuals and obese patients (Caro et al. 1996), the former show a four fold increase in the ratio, thus suggesting altered leptin penetrance into the CSF in obese patients. Such individual differences in leptin penetrance into the CSF could serve as an explanation as to why OHS only develops in certain obese patients with OSA (Yee et al. 2006).
4. Clinical consequences of OHS
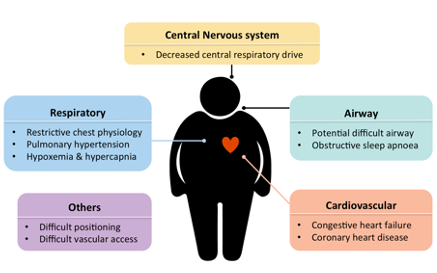
Figure 3 | Possible clinical manifestations of patients with obesity hypoventilation syndrome (Adapted from Mokhlesi et al. 2012)
Morbid obesity and OSAS often result in disturbed PaO2 and PaCO2, however, OHS patients exhibit an exaggerated lower daytime PaO2 and higher Paco2. These patients spend a great proportion of their sleep at oxygen saturation of no more than 90%. This in turn can lead to pulmonary hypertension, peripheral oedema and cor pulmonale (Flegal et al. 2005).
Due to the similarities between OHS and OSAS, patients suffering from the former are often overlooked within groups of patients present in sleep clinics or those hospitalised for acute illness or chronic stable disease (Punjabi et al. 2009). Considering that healthcare costs associated with OHS patients are significantly higher (Marin et al. 2005); the combination of late diagnosis and high costs of this condition impose a great burden to both the patient and the healthcare system.
Furthermore, cardio-metabolic morbidity is another complication that occurs more commonly in patients with OHS than those with OSAS and extreme obesity. When comparing eucapnic obese patients with OHS individuals, the latter suffer from higher rates of systemic hypertension, congestive and right heart failure as well as angina (FIG. 3) (Hida 2003). In addition, higher insulin resistance and more frequent need for glucose controlling medication is another characteristic of OHS (Marin et al. 2005). Considering the relatively well-studied link between OSA, obesity and cardio-metabolic disease, such findings in OHS patients are not unexpected.
Data from investigations into the molecular basis of OHS reveal an upregulation in expression of proatherogenic chemokines, reduced adiponectin levels (an antiatherogenic adipokine) and compromised endothelial function (Golpe 2002). Furthermore, the persistence of high inflammatory markers in OHS patients, both before and after treatment for disordered breathing is another influence believed to negatively impact the prognosis of patients in this category (Kessler et al. 1996).
OHS patients are subject to various lifestyle limitations. Dyspnoea and sleepiness are manifests of the respiratory and obesity aspect of OHS that severely impact the mental and physical wellbeing of these patients, thus negatively affecting quality of life (Lin et al. 2004).
A significant number of severely obese OHS patients who undergo treatment for improving gas exchange continue to score low on the quality of life questionnaire (Nowbar et al. 2004). On the other hand, another study carried out in Japan on OHS patients with moderate obesity reports of marked improvement in their overall quality of life after a 3 to 6 month course of CPAP therapy (Sugerman et al. 1988). This difference is most likely a reflection of the ongoing detrimental effects of severe obesity on health and wellbeing. Quality of life is a key issue in patients with hypoventilation, as there appears to be a strong link between mortality and health related quality of life in this subset of patients.
5. OHS screening and identification
Elevated awake PaCO2 is a diagnostic marker of OHS, which requires routine testing of arterial blood gases. One of the challenges in identifying chronic OHS is its similar presentation to uncomplicated OSAS, thus increasing the risk of the condition being overlooked.
OHS screening involving readily available tests such as pulse oximetry and serum bicarbonate measurements can be relatively inexpensive and useful methods for identifying susceptible individuals (Mandal et al. 2014). Increased CO2levels can be detected with a high sensitivity (92%) albeit low specificity (50%) in individuals with a serum bicarbonate of 27 mEq/L or greater (Monneret 2015). In addition, most OHS patients have a PaO2 of less than 70 mm Hg, a figure that is more prominent in advanced OHS (Mokhlesi et al. 2012). In order to achieve a more accurate diagnosis, there is a need for measurement of arterial blood gasses to accompany pulse oximetry readings that indicate a saturation of less than 94%.
A comprehensive polysomnography test can be an expensive investigation in many countries, thus there has been a surge in popularity of simplified monitoring devices that help in diagnosis of OSAS. In lieu of this, the use of simplified auto titrating pressure devices has also been inclining. Unattended titration of CPAP in OHS is against the current clinical guidelines; however, since OHS can be overlooked, it is common for a percentage of individuals to be incorrectly treated as such (Morgenthaler et al. 2008). However, there are conflicting data regarding the effectiveness of CPAP in OHS, whereby a proportion of patients respond positively to CPAP (Mokhlesi et al. 2012), whereas others experience deteriorating conditions as a result of persistent nocturnal desaturation and hypoventilation (Banerjee et al. 2007). This highlights the importance of correct diagnosis of OHS prior to commencing treatment.
Furthermore, transcutaneous CO2 measurements can aid in measurement of CO2 retention particularly during REM sleep and help in guiding the titration of PAP therapy for adequate ventilation (Mandal et al. 2014; Maniscalco et al. 2008).
6. Treatment
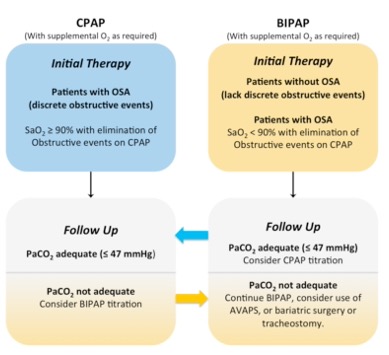
Figure 4 | A schematic summary portraying the proposed criteria for use of CPAP or BIPAP therapy in patients with OHS. BIPAP has been recommended for those who continue to have oxygen saturations (SaO2) of less than 90% with elimination of obstructive events. (Adapted from Ojeda et al. 2015)
6.1 Positive Airway Pressure Therapy
In 1982, Rapoport et al. first described the use of continuous positive airway pressure (CPAP) therapy in management of OHS. Since, numerous studies have been conducted attesting to its efficacy. However, in certain individuals CPAP fails to achieve progressive results, this alone has lead to investigations as to whether alternatives such as bi-level PAP (a flow generator with distinct pressures for inhalation and expiration) therapy could be more beneficial in management of OHS. (Rapoport et al. 1982)
Further investigations into the efficacy of CPAP in OHS, in context of a prospective study has shown that in a group of patients with severe OHS, just over half the cohort (57%) show successful titration with a mean pressure of 13.9 cm/H2O using CPAP alone; whereas the remaining individuals continued to display hypoxemia at therapeutic (or near therapeutic) pressures with approximately 20% of their total sleep time spent at oxygen saturations of below 90% (Banerjee et al. 2007). In addition, obesity was detected to be more prominent in patients who didn’t show ideal response to CPAP. Furthermore, the CPAP non-responders in this group had residual OSA (AHI of 25 events/h), which may be an indication of CPAP failure to reach therapeutic pressures.
When comparing the long-term efficacy of bi-level PAP versus CPAP in OHS, a prospective randomised study has shown that bi-level PAP is not always necessarily the superior option over CPAP (Piper et al. 2008). The decision as to which mode of treatment to use is more likely a case of individualising the therapy based on disease severity and patient response to each mode of treatment, rather than always using the most advanced and complex titration devices. The same study showed that as long as there is effective CPAP therapy for OSA and nocturnal hypoxemia, there was no apparent difference in OHS management using CPAP or bi-level PAP over a course of 3 months, whereby 80% of the OHS patients in the study were titrated successfully with CPAP alone.
In situations where OHS patients with unresolved apnoea and hypopnoea display intolerance towards high CPAP pressures (15 cm/H2O or more), then the use of bi-level PAP is recommended to achieve successful titration (Kushida et al. 2008). Furthermore, there is evidence that average volume assured pressure support (AVAPS) bi-level PAP ventilators can provide superior control of nocturnal sleep disordered breathing (Janssens et al. 2009), however, a recent randomised control trial has shown that there is no significant difference in treatment outcome between the two modes (Murphy et al. 2012).
6.2 Oxygen therapy
To maintain a SpO2 of 90% and above in OHS patients, nearly 50% require oxygen therapy (Kaw et al. 2009). A cohort study has shown that adherence to PAP therapy reduces the need for daytime oxygen therapy from 30% to 6% (Banerjee et al. 2007). Furthermore, it has been shown that hypercapnia can be exacerbated with moderate concentrations of supplemental oxygen in OHS (Hollier et al. 2013). Therefore, after a period of time it is necessary for patients on PAP therapy to be re-evaluated for their oxygen requirements both nocturnally and diurnally.
6.3 Tracheostomy
Whilst nowadays tracheostomy is an option largely reserved for OHS patients who do not benefit from PAP therapy or show signs of cor pulmonale, it was the first mode of treatment described for management of OHS (Hensley & Read 1976). A retrospective study has shown that tracheostomy is linked to marked improvements in OHS in terms of reduced events of mean non-rapid-eye-movement (non-REM) AHI and REM AHI per hour. Individuals, who do not see a short-term significant improvement after tracheostomy, go on to have marked improvements in the severity of their sleep disordered breathing in the long term, owing to improvements in their hypercapnia.
6.4 Bariatric surgery
There have been contrasting data emerging from studies focusing on the long-term benefits of bariatric surgery in OSA. A study has shown that despite significant reduction in AHI of patients with severe OSA who had undergone bariatric surgery, some continued to have moderate OSA and thus were indicated to continue PAP therapy for reduction in sleep disordered breathing (Haines et al. 2007). In addition, a similar study reported of development of severe OSA 7 years post operatively in patients with mild OSA who had undergone bariatric surgery. In addition, a meta-analysis of 12 studies with a cohort of 342 patients showed that although bariatric surgery leads to significant reduction in the AHI of OSA patients, a large number of patients (62%) continue to have residual disease (Pillar et al. 1994) (Borel et al. 2013). Weight increase and recurrence of sleep-disordered breathing post bariatric surgery is a common occurrence and thus such patients must be observed regularly.
With regards to the impact of bariatric surgery on OHS, a study has shown that after surgery patients displayed initial improvements in their PaO2 and PaCO2 levels, however, over a period of five years, these individuals went on to have worsened PaO2 and PaCO2 with no significant increase in their BMI (Sugerman et al. 1992). It is suggested that these results are due to re-development of disordered breathing leading to deteriorating levels of daytime blood gases.
6.5 Pharmacological Respiratory Stimulation
Medroxyprogesterone and Acetazolamide are both respiratory stimulants that act on the hypothalamus and the metabolism respectively. The use of Medroxyprogesterone in OHS patients who can normalise their PaCO2 with voluntary hyperventilation (i.e. have no mechanical impairment) is associated with reduced PaCO2 and raised PaO2 (Douketis et al. 2005; Bayliss & Millhorn 1992). However, there are mixed reports concerning the efficacy of this treatment; nevertheless, careful assessment is key before administration of medroxyprogesterone, since it is associated with increased risk of venous thromboembolism and breakthrough uterine bleeding in women (Bayliss & Millhorn 1992).
The inhibition of carbonic anhydrase induces metabolic acidosis that leads to increase minute ventilation. This is the basis of Acetazolamide’s mechanism of action. There is limited data on the effectiveness of this drug on OHS, but amongst this limited pool of data, acetazolamide has been shown to reduce AHI in patients with OSA of moderate to severe severity, albeit with varying efficacy and side effects (Tojima et al. 1988).
Ultimately, PAP remains the most studied and commonly used therapy in management of OHS. Other modes of therapy gain importance in cases where individuals are unresponsive to PAP, whereby there is a need for alternative treatments that utilise a mixture of PAP, pharmacotherapy or surgical approaches.
7. Summary & future directions
The current trend of increasing obesity is expected to trigger a surge in prevalence of OHS. The ominous combination of late diagnosis and high morbidity associated with OHS emphasises the need for reliable methods of early diagnosis and formulation of appropriate methods of therapy. Whilst PAP therapy is associated with a positive outcome in majority of OHS patients, there remain individuals who are unresponsive to this mode of treatment, and thus other options such as bariatric surgery, tracheostomy or pharmacological respiratory stimulants must be further studied.
Improving patient adherence to PAP therapy and counselling on weight reduction can be effective methods for current management of OHS. Future studies will undoubtedly pave the way for new discoveries in the pathophysiology of OHS and help in emergence of novel and effective methods of therapy.
References
Obesity, a major public health concern, increasingly impacts key physiological systems, including respiratory health.Akashiba, T. et al., 2006. Clinical Characteristics of Obesity-hypoventilation Syndrome in Japan: a Multi-center Study. Internal Medicine, 45(20), pp.1121–1125.
Banerjee, D. et al., 2007. Obesity Hypoventilation Syndrome: Hypoxemia During Continuous Positive Airway Pressure. CHEST Journal, 131(6), pp.1678–1684.
Bayliss, D.A. & Millhorn, D.E., 1992. Central neural mechanisms of progesterone action: application to the respiratory system. Journal of Applied Physiology, 73(2), pp.393–404.
BECKER, H.F. et al., 1999. Breathing during Sleep in Patients with Nocturnal Desaturation. American Journal of Respiratory and Critical Care Medicine, 159(1), pp.112–118.
Borel, J.-C. et al., 2013. Comorbidities and Mortality in Hypercapnic Obese under Domiciliary Noninvasive Ventilation X.-L. Ma, ed. PLOS ONE, 8(1), p.e52006.
Caro, J.F. et al., 1996. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. The Lancet, 348(9021), pp.159–161.
CHERNIACK, N.S. et al., 2008. Effect of Serum Leptin Levels on Hypercapnic Ventilatory Response in Obstructive Sleep Apnea. Respiration, 75(3).
De Llano, L.A.P. et al., 2005. Short-term and Long-term Effects of Nasal Intermittent Positive Pressure Ventilation in Patients With Obesity-Hypoventilation Syndrome. CHEST Journal, 128(2), pp.587–594.
Douketis, J.D. et al., 2005. Does the type of hormone replacement therapy influence the risk of deep vein thrombosis? A prospective case–control study. Journal of Thrombosis and Haemostasis, 3(5), pp.943–948.
Ojeda, O.C. et al., 2015. Noninvasive mechanical ventilation in patients with obesity hypoventilation syndrome. Long-term outcome and prognostic factors. Archivos de bronconeumologia, 51(2), pp.61–68.
Emirgil, C. & Sobol, B.J., 2015. The Effects of Weight Reduction on Pulmonary Function and the Sensitivity of the Respiratory Center in Obesity1, 2. American Review of Respiratory Disease.
Finucane, M.M. et al., 2011. National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9·1 million participants. The Lancet, 377(9765), pp.557–567.
Flegal, K.M. et al., 2005. Excess Deaths Associated With Underweight, Overweight, and Obesity. JAMA, 293(15), pp.1861–1867.
Freedman, D.S., 2002. Trends and Correlates of Class 3 Obesity in the United States From 1990 Through 2000. JAMA, 288(14), pp.1758–1761.
Golpe, R., 2002. Diurnal Hypercapnia in Patients With Obstructive Sleep Apnea Syndrome. CHEST Journal, 122(3), pp.1100–1101.
Haines, K.L. et al., 2007. Objective evidence that bariatric surgery improves obesity-related obstructive sleep apnea. Surgery, 141(3), pp.354–358.
Han, F. et al., 2001. Treatment Effects on Carbon Dioxide Retention in Patients With Obstructive Sleep Apnea-Hypopnea Syndrome. CHEST Journal, 119(6),
pp.1814–1819.
Hart, N. et al., 2013. Obesity hypoventilation syndrome: does the current definition need revisiting? Thorax, 69(1), pp.thoraxjnl–2013–204298–84.
Hensley, M.J. & Read, D.J.C., 1976. Intermittent Obstruction of the Upper Airway during Sleep Causing Profound Hypoxaemia. A Neglected Mechanism Exacerbating Chronic Respiratory Failure. Australian and New Zealand Journal of Medicine, 6(5), pp.481–486.
Hida, W., 2003. Quality of Life in Obesity Hypoventilation Syndrome. Sleep and Breathing, 7(1), pp.1–2.
Hollier, C.A. et al., 2013. Moderate concentrations of supplemental oxygen worsen hypercapnia in obesity hypoventilation syndrome: a randomised crossover study. Thorax, 69(4), pp.thoraxjnl–2013–204389–353.
Janssens, J.-P. et al., 2003. Changing Patterns in Long-term Noninvasive Ventilation: A 7-Year Prospective Study in the Geneva Lake Area. CHEST Journal, 123(1), pp.67–79.
Janssens, J.-P., Metzger, M. & Sforza, E., 2009. Impact of volume targeting on efficacy of bi-level non-invasive ventilation and sleep in obesity-hypoventilation. Respiratory medicine, 103(2), pp.165–172.
Jokic, R. et al., 2000. Ventilatory responses to hypercapnia and hypoxia in relatives of patients with the obesity hypoventilation syndrome. Thorax, 55(11), pp.940–945.
Kaw, R. et al., 2009. Determinants of Hypercapnia in Obese Patients With Obstructive Sleep Apnea: A Systematic Review and Metaanalysis of Cohort Studies. CHEST Journal, 136(3), pp.787–796.
Kessler, R. et al., 1996. Pulmonary hypertension in the obstructive sleep apnoea syndrome: prevalence, causes and therapeutic consequences. European Respiratory Journal, 9(4), pp.787–794.
Kushida, C.A. et al., 2008. Clinical guidelines for the manual titration of positive airway pressure in patients with obstructive sleep apnea. Journal of clinical sleep medicine : JCSM : official publication of the American Academy of Sleep Medicine, 4(2), pp.157–171.
Laaban, J.-P. & Chailleux, E., 2005. Daytime Hypercapnia in Adult Patients With Obstructive Sleep Apnea Syndrome in France, Before Initiating Nocturnal Nasal Continuous Positive Airway Pressure Therapy. CHEST Journal, 127(3), pp.710–715.
Laila Al Dabal, A.S.B., 2005. Obesity hypoventilation syndrome. Annals of Thoracic Medicine, 4(2), pp.41–49.
Lavie, P., 2008. Who was the first to use the term Pickwickian in connection with sleepy patients? History of sleep apnoea syndrome. Sleep medicine reviews, 12(1), pp.5–17.
Leech, J. et al., 1991. Voluntary hyperventilation in obesity hypoventilation. CHEST Journal, 100(5), pp.1334–1338.
Lin, C.-C. et al., 2004. Oral airway resistance during wakefulness in eucapnic and hypercapnic sleep apnea syndrome. Respiratory Physiology & Neurobiology, 139(2), pp.215–224.
Lin, C.C., 1994. Effect of nasal CPAP on ventilatory drive in normocapnic and hypercapnic patients with obstructive sleep apnoea syndrome. European Respiratory Journal, 7(11), pp.2005–2010.
Mandal, S. et al., 2014. A cohort study to identify simple clinical tests for chronic respiratory failure in obese patients with sleep-disordered breathing. BMJ Open Respiratory Research, 1(1), pp.e000022–e000022.
Maniscalco, M. et al., 2008. Evaluation of a transcutaneous carbon dioxide monitor in severe obesity. Intensive Care Medicine, 34(7), pp.1340–1344.
Marin, J.M. et al., 2005. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. The Lancet, 365(9464), pp.1046–1053.
Masa, J.F. et al., 2001. The Obesity Hypoventilation Syndrome Can Be Treated With Noninvasive Mechanical Ventilation. CHEST Journal, 119(4), pp.1102–1107.
Mokhlesi, B., 2010. Obesity Hypoventilation Syndrome: A State-of-the-Art Review. Respiratory Care, 55(10), pp.1347–1365.
Mokhlesi, B. et al., 2006. Impact of adherence with positive airway pressure therapy on hypercapnia in obstructive sleep apnea. Journal of clinical sleep medicine : JCSM : official publication of the American Academy of Sleep Medicine, 2(1), p.57.
Mokhlesi, B. et al., 2007. Obesity hypoventilation syndrome: prevalence and predictors in patients with obstructive sleep apnea. Sleep and Breathing, 11(2), pp.117–124.
Mokhlesi, B., Kryger, M.H. & Grunstein, R.R., 2012. Assessment and Management of Patients with Obesity Hypoventilation Syndrome. Proceedings of the American Thoracic Society, 5(2), pp.218–225.
Monneret, D., 2015. Bicarbonate or Base Excess in Early Obesity Hypoventilation Syndrome: A Methodologic Viewpoint. CHEST Journal, 147(6), pp.e231–e231.
Morgenthaler, T.I. et al., 2008. Practice Parameters for the Use of Autotitrating Continuous Positive Airway Pressure Devices for Titrating Pressures and Treating Adult Patients with Obstructive Sleep Apnea Syndrome: An Update for 2007: An American Academy of Sleep Medicine Report. Sleep, 31(1), pp.141–147.
Murphy, P.B. et al., 2012. Volume targeted versus pressure support non-invasive ventilation in patients with super obesity and chronic respiratory failure: a randomised controlled trial. Thorax, 67(8), pp.727–734.
Nowbar, S. et al., 2004. Obesity-Associated hypoventilation in hospitalized patients: prevalence, effects, and outcome. The American Journal of Medicine, 116(1), pp.1–7.
Phipps, P.R. et al., 2002. Association of serum leptin with hypoventilation in human obesity. Thorax, 57(1), pp.75–76.
Pillar, G., Peled, R. & Lavie, P., 1994. Recurrence of Sleep Apnea Without Concomitant Weight Increase 7.5 Years After Weight Reduction Surgery. CHEST Journal, 106(6), pp.1702–1704.
Piper, A.J. & Grunstein, R.R., 2010. Big breathing: the complex interaction of obesity, hypoventilation, weight loss, and respiratory function. Journal of Applied Physiology, 108(1), pp.199–205.
Piper, A.J. et al., 2008. Randomised trial of CPAP vs bilevel support in the treatment of Obesity Hypoventilation Syndrome without severe nocturnal desaturation. Thorax, 63(5), pp.395–401.
Punjabi, N.M. et al., 2009. Sleep-Disordered Breathing and Mortality: A Prospective Cohort Study A. Patel, ed. PLOS Med, 6(8), p.e1000132.
Quint, J.K., Ward, L. & Davison, A.G., 2007. Previously undiagnosed obesity hypoventilation syndrome. Thorax, 62(5), pp.462–463.
Rapoport, D.M. et al., 1982. Reversal of the Pickwickian Syndrome by Long-Term Use of Nocturnal Nasal-Airway Pressure. New England Journal of Medicine, 307(15), pp.931–933.
Resta, O. et al., 2000. Prevalence and mechanisms of diurnal hypercapnia in a sample of morbidly obese subjects with obstructive sleep apnoea. Respiratory medicine, 94(3), pp.240–246.
Sakakibara, H. et al., 1999. Cephalometric abnormalities in non‐obese and obese patients with obstructive sleep apnoea. European Respiratory Journal, 13(2), pp.403–410.
Salord, N. et al., 2013. Continuous positive airway pressure in clinically stable patients with mild‐to‐moderate obesity hypoventilation syndrome and obstructive sleep apnoea. Respirology, 18(7), pp.1135–1142.
Sidney Burwell, C. et al., 1956. Extreme obesity associated with alveolar hypoventilation—A pickwickian syndrome. The American Journal of Medicine, 21(5), pp.811–818.
Society, A.T., 2002. ATS/ERS statement on respiratory muscle testing. Am J Respir Crit Care Med, 166(4), pp.518–624.
Sotos, J.G., 2003. Taft and Pickwick: Sleep Apnea in the White House. CHEST Journal, 124(3), pp.1133–1142.
Spritzer, D.A., 2004. Global Health: Obesity epidemic migrates east. CMAJ : Canadian Medical Association Journal, 171(10), pp.1159–1159.
Sugerman, H.J. et al., 1988. Hemodynamic dysfunction in obesity hypoventilation syndrome and the effects of treatment with surgically induced weight loss. Annals of Surgery, 207(5), pp.604–613.
Sugerman, H.J. et al., 1992. Long-term effects of gastric surgery for treating respiratory insufficiency of obesity. The American Journal of Clinical Nutrition, 55(2 Suppl), pp.597S–601S.
Tankersley, C.G. et al., 1998. Leptin attenuates respiratory complications associated with the obese phenotype. Journal of Applied Physiology, 85(6), pp.2261–2269.
Teichtahl, H., 2001. The Obesity-Hypoventilation Syndrome Revisited. CHEST Journal, 120(2), pp.336–339.
Tojima, H. et al., 1988. Effects of acetazolamide in patients with the sleep apnoea syndrome. Thorax, 43(2), pp.113–119.
Weatherall, M. & Beasley, R., 2011. The Effect of Supplemental Oxygen on Hypercapnia in Subjects With Obesity-Associated Hypoventilation. CHEST.
Yee, B.J. et al., 2006. Treatment of obesity hypoventilation syndrome and serum leptin. Respiration, 73(2), pp.209–212.




Leave a Comment